








公众号/
编辑 | 萝卜皮
蛋白质设计一直依赖于结构功能与蛋白序列的对应关系。科学家一直希望解决从骨架原子坐标逆向预测蛋白质序列的问题。迄今为止,解决该问题的机器学习方法均受到可用实验确定的蛋白质结构数量的限制。
加州大学伯克利分校和 Facebook 人工智能团队的研究人员,通过使用 AlphaFold2 预测 12M 蛋白质序列的结构,将训练数据增加了近三个数量级。使用这些额外数据进行训练,具有不变几何输入处理层的序列到序列转换器在结构上保持的主干上实现了 51% 的本地序列恢复,对于掩埋残基的恢复率为 72%,总体上比现有方法提高了近 10 个百分点 。
该模型可推广完成各种更复杂的任务,包括蛋白质复合物的设计、部分掩蔽结构、结合界面和多种状态。
该研究以「Learning inverse folding from millions of predicted structures」为题,于 2022 年 4 月 10 日发布在 BioRxiv 预印平台。

设计编码具有所需特性的蛋白质的新氨基酸序列,称为从头蛋白质设计,是生物工程的核心挑战。解决这个问题的最成熟的方法使用能量函数,它直接模拟蛋白质折叠状态的物理基础。
近期,科学家们已经提出了一类新的基于深度学习的方法,使用生成模型来预测结构的序列,生成主干结构,联合生成结构和序列,或直接建模序列。直接从数据中学习蛋白质设计规则的潜力使深度生成模型,成为当前基于物理的能量函数的最有希望的替代方案。

图示:用预测的结构增强逆折叠。(来源:论文)
然而,实验确定的蛋白质结构数量相对较少,这限制了深度学习方法。实验确定的结构覆盖不到 0.1% 的已知蛋白质序列空间。而 UniRef 序列数据库有超过 5000 万个簇,序列同一性为 50%;截至 2022 年 1 月,蛋白质数据库 (PDB) 包含少于 53,000 个独特序列的结构,这些序列以相同的同一性水平聚集。

图示:所考虑的蛋白质设计任务的说明。(来源:论文)
在这里,Facebook 人工智能团队的研究人员探讨了,是否可以使用预测结构来克服实验数据的限制。随着蛋白质结构预测的进展,现在可以考虑从大规模预测结构中学习。预测大型数据库中序列的结构可以将蛋白质序列的结构覆盖范围扩大几个数量级。为了训练蛋白质设计的逆模型,研究人员使用 AlphaFold2 预测 UniRef50 中 1200 万个序列的结构。
从骨架结构逆向预测序列的问题,被称为反向折叠或固定骨架设计。他们使用自回归编码器-解码器架构,将逆折叠作为序列到序列问题来处理,其中模型的任务是从其骨架原子的坐标中恢复蛋白质的天然序列。
研究人员将大量具有未知结构的序列,添加为额外的训练数据;当实验结构未知时,根据预测结构来调整模型。这种方法与机器翻译中的反向翻译平行,其中一个方向的预测翻译用于改进相反方向的模型。即使预测的输入(即结构)质量低下,反向翻译也能有效地从额外的目标数据(即序列)中学习。
研究人员发现现有方法受到数据的限制。虽然当前最先进的逆折叠模型在训练中使用预测结构时会退化,但更大的模型和不同的模型结构可以有效地从额外的数据中学习,从而使结构上伸出的原生主干的序列恢复率提高近 10 个百分点。
他们根据先前工作中的固定主干设计基准来评估模型,并评估一系列任务的泛化能力,包括复合物和结合位点的设计、部分掩蔽的主干和多个构象。研究人员进一步考虑使用模型来零样本预测突变对蛋白质功能和稳定性、复杂稳定性和结合亲和力的影响。
固定骨架蛋白设计
研究人员从给定骨架原子(N、Cα、C)坐标的情况下预测天然蛋白质序列的任务开始。保留原生序列的困惑度和序列恢复是此任务的两个常用指标。困惑度测量预测序列分布中天然序列的逆可能性(高可能性的低困惑度)。序列恢复(准确度)测量采样序列与每个位置的天然序列匹配的频率。为了最大限度地恢复序列,预测的序列是在低温 T = 1e-6 的情况下从模型中采样的。下表比较了使用这些指标在结构上保持的主干上的模型。
表:固定骨架序列设计。

从结果可以观察到,当前最先进的逆折叠模型受到 CATH 训练集的限制。在 CATH 数据集上将当前的 1M 参数模型 (GVP-GNN) 缩放到 21M 参数 (GVP-GNN-large) 会导致过度拟合,序列恢复率从 42.2% 下降到 39.2%。
另一方面,1M 参数规模的当前模型无法利用预测结构:使用预测结构训练 GVP-GNN 会导致序列恢复率下降到 38.6%,随着训练中预测结构数量的增加,性能会恶化。
较大的模型受益于 AlphaFold2 预测的 UniRef50 结构的训练。使用预测结构进行训练将 GVP-GNN-large 的序列恢复率从 39.2% 提高到 50.8%,将 GVP-Transformer 的序列恢复率从 38.3% 提高到 51.6%,而且这仅仅是在实验派生结构上进行训练。
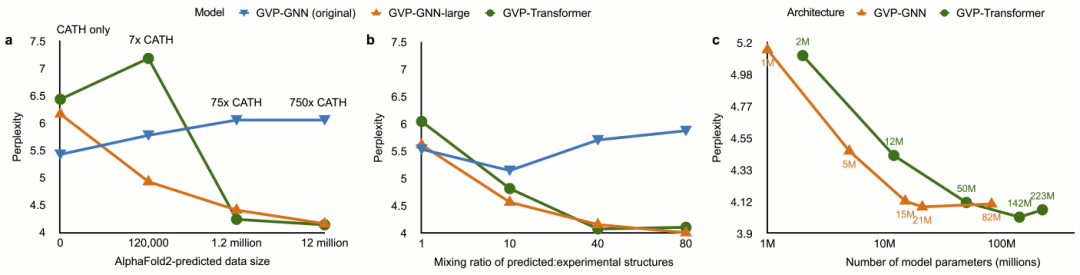
图示:训练数据的消融研究。(来源:论文)
之后研究人员还在「部分掩蔽的主干」、「蛋白复合物」、「多种构象」等多个方面对模型进行了测试与评估,均得到了不错的效果。
零样本预测
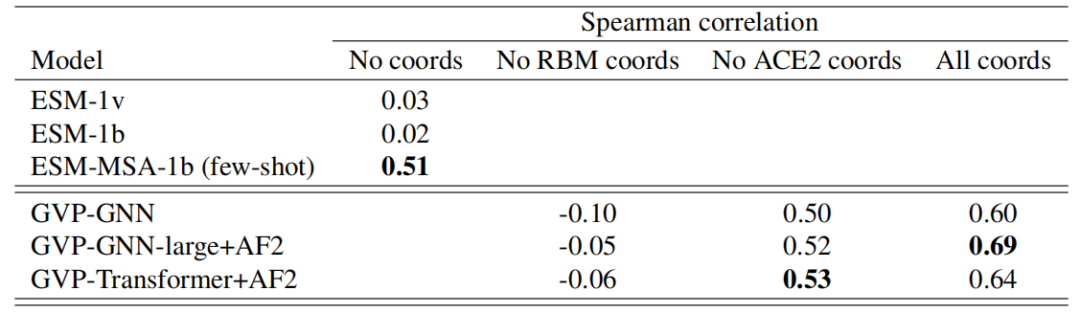
之后的研究表明,逆折叠模型可以作为实际设计应用中突变效应的有效零样本预测因子,包括预测复杂稳定性、结合亲和力和插入效应。
为了对突变对特定序列的影响进行评分,在给定实验确定的野生型结构的情况下,研究人员根据反向折叠模型使用突变和野生型序列的可能性之间的比率。GVP-GNN 和 GVP-Transformer 都可以进行精确的似然评估,因为它们都基于自回归解码器。然后,研究人员将这些似然比分数与在同一组序列上测量的实验确定的适应度值进行比较。
表:基于 ACE2-RBD 突变扫描数据评估的 SARS-CoV-2 Spike 受体结合域 (RBD) 结合亲和力预测的零样本性能。
该团队的研究还对「基于结构的蛋白质序列设计」、「蛋白质的生成模型」、「语言模型」、「结构不可知的蛋白质序列设计」、「回译」等领域的研究工作带来了重要启发。
结语
虽然最大的序列数据库中有数十亿个蛋白质序列,但可用的实验确定结构的数量约为数十万,这对从蛋白质结构数据中学习的生成方法施加了限制。在这项研究中,研究人员探讨了最近深度学习方法的预测结构是否可以与实验结构一起用于训练蛋白质设计模型。
为此,他们使用 AlphaFold2 为 1200 万个 UniRef50 序列生成了结构。作为使用这些数据进行训练的结果,研究人员观察到困惑度和序列恢复的显着提高,并证明了对更长蛋白质复合物、多种构象的蛋白质以及对结合亲和力和 AAV 包装的突变影响的零样本预测的泛化。
这些结果突出表明,除了几何归纳偏差是迄今为止反折叠工作的主要重点之外,寻找利用更多训练数据源的方法是提高建模能力的同样重要的途径。
他们还朝着更一般的结构条件蛋白质设计任务迈出了第一步。通过将主干跨度掩蔽集成到反向折叠任务中,并使用序列到序列转换器,可以对短掩蔽跨度进行合理的序列预测。
如果可以找到方法继续利用预测的结构来构建蛋白质的生成模型,那么就有可能创建模型,来学习从结构目前未知的数十亿自然序列的扩展宇宙中设计蛋白质。
论文链接:https://www.biorxiv.org/content/10.1101/2022.04.10.487779v1

 友情链接
友情链接