








公众号/
编辑 | 萝卜皮
生物技术行业一直在寻找完美的突变,将不同蛋白质的特性合成组合以达到预期的效果。可能有必要开发新的药物或酶来延长酸奶的保质期,在野外分解塑料,或使洗衣粉在低水温下有效。
蛋白质序列的学习表示可以大大改进进行生物学预测的系统,并且还可能有助于揭示以前未发现的生物学信息。如何选择表示数据,对后续从中提取信息的能力有根本性的影响。
机器学习有望从大型非结构化数据集(例如生物学中出现的数据集)中自动确定有效表示。然而,经验证据表明,这些机器学习模型看似微小的变化会产生截然不同的数据表示,从而导致对数据的不同生物学解释。这就引出了一个问题,即什么才是最有意义的表示。
丹麦技术大学和哥本哈根大学的研究人员研究了这个问题,并探索了表示蛋白质序列的方法。他们主要探讨了自然产生表征的两个关键上下文(context):迁移学习和可解释学习。
在迁移学习上下文中,研究人员证明了几种当代实践产生了次优的性能;在可解释学习中,他们证明将表示几何考虑在内可显著提高可解释性,并使用模型揭示了之前方法被掩盖的生物信息。
该研究以「Learning meaningful representations of protein sequences」为题,于 2022 年 4 月 8 日发布在《Nature Communications》。

数据表示在生物数据的统计分析中起着至关重要的作用。从本质上讲,表示是将原始数据提炼成一个抽象的、高级的、通常是低维的空间,该空间捕获原始数据的基本特征。随后可以将其用于数据探索,例如通过可视化,或在可用数据有限的情况下进行特定任务的预测。
鉴于表示的重要性,说明生物学表示学习(表征)的兴起并不奇怪,这是机器学习的一个子领域,其中表示与统计模型一起估计。特别是在蛋白质序列的分析中,过去几年已经产生了许多研究,说明表征对于「如何从通过测序技术获得的数百万数据中自动提取关键信息」十分重要。
图示:蛋白质序列的表示。(来源:论文)
虽然这些有希望的结果表明,表示学习可以对科学数据分析产生重大影响,但它们也带来了一个问题:什么是好的表示?
表示学习的一个经典例子是主成分分析(PCA),它学习与原始数据线性相关的特征。当代技术消除了线性假设,转而寻求高度非线性关系,通常通过使用神经网络。
这在自然语言处理(NLP)中特别成功,其中单词序列的表示是从大量在线文本资源中学习的,提取支持后续特定语言任务的语言的一般属性。这种词序列模型的成功激发了其在生物序列建模中的应用,在远程同源物检测、功能分类和突变效应预测等应用领域取得了令人瞩目的成果。
由于表征正在成为生物序列分析的重要组成部分,应该批判性地思考所构建的表征是否有效地捕获了真正想要的信息。研究人员讨论了这个主题,重点是蛋白质序列,尽管许多见解也适用于其他生物序列。该工作由两部分组成。
首先,考虑迁移学习设置中的表示;他们调查了网络设计和训练协议对结果表示的影响,并发现当前的几种做法不是最理想的。
其次,研究了出于数据解释目的而使用表示;结果表明,表示几何的显式建模使研究人员能够提取稳健且可识别的生物学结论。
该研究的结论阐明了,「什么构成蛋白质的有意义表示」问题的部分答案。结论之一是问题本身没有一个单一的通用答案,并且必须始终通过对表示目的的规范进行限定。
适合进行预测的表示,可能不是人类研究人员更好地理解潜在生物学的最佳选择,反之亦然。因此,所有任务的单一蛋白质表示的诱人想法在实践中似乎是行不通的。
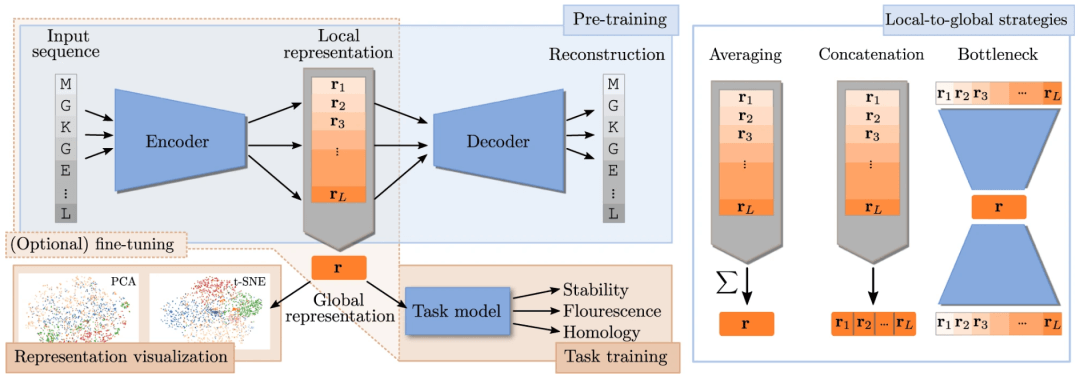
设计有目的的表示
为给定任务设计表示需要反思研究者希望表示封装哪些生物学特性。蛋白质的不同生物学方面将对表示提出不同的要求,但在表示中强制执行特定属性并不简单。然而,研究人员可以通过
(1)选择合适的模型架构;
(2)预处理数据;
(3)选择合适的目标函数;
(4)将先验分布放在模型的某些部分上来引导表示学习。
知情的网络架构可能难以构建,因为通常的神经网络「构建块」是相当基本的数学函数,不会立即与高级生物信息相关联。尽管如此,对长度不变序列表示的讨论是一个简单的例子,说明了人们如何为任务生物学的模型架构提供信息。人们普遍认为,全局蛋白质特性与局部特性不是线性相关的。
因此,当允许模型学习这种非线性关系而不是依赖于局部表示的公共线性平均值时,模型性能显著提高也就不足为奇了。将这个想法推广到这里探索的 Resnet 架构之外会很有趣,特别是结合最近的大规模基于转换器的语言模型。
研究人员推测,虽然类似的「唾手可得的果实」可能仍然存在于当前应用的网络架构中,但它们是有限的,需要更先进的工具将生物信息编码到网络架构中。基于注意力的架构中的内部表示,已被证明可以恢复蛋白质之间已知的物理相互作用,从而为整合有关蛋白质中已知物理相互作用的先验信息打开了大门。近期关于神经网络中排列和旋转不变性/等变性的研究工作均很有希望,尽管它们尚未在表示学习中进行详尽的探索。

图示:编码到潜在表示空间中的系统发育树。(来源:论文)
数据预处理和特征工程在当代「端到端」表示学习中不受欢迎,但它仍然是模型设计的重要组成部分。特别是,使用来自计算生物学的大量现有工具进行预处理,是将现有生物学知识编码到表示中的一种有价值的方法。
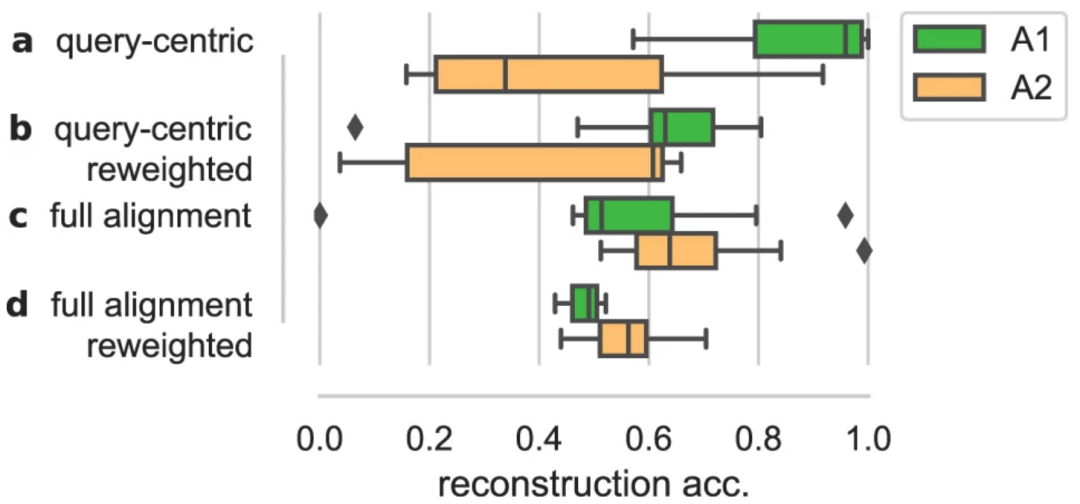
图示:对齐预处理对表示可靠地解码回蛋白质序列的能力的影响。(来源:论文)
在对对齐的蛋白质序列进行训练时,可以看到无监督模型的表示能力有了显著提高,因为这注入了关于一组序列中可比较序列位置的先验知识。虽然近期的研究工作越来越多地致力于直接从数据中学习此类信号的技术,但仍不清楚多重对齐所提供的优势是否可以被这些方法完全代替。
其他预处理技术,例如序列的重新加权,目前也依赖于对齐序列。这些例子表明,如果学界向「端到端」学习迈进得太快,可能会因为放弃现有工具中多年的经验,而出现将婴儿和洗澡水一起扔出去的荒诞事件。
相关的目标函数对于任何学习任务都至关重要。尽管表示学习通常使用重建损失进行,但研究人员证明根据此目标的最佳表示对于任何特定的迁移学习任务通常都不是最佳的。这表明应该根据下游任务特定的性能来选择表示的超参数,而不是在保留集上的重建性能。
然而,这是一个微妙的过程,因为在下游任务上优化表示模型的参数与过度拟合的高风险有关。研究人员预计,将大型无监督数据集上的重建目标与半监督学习环境中的特定任务目标相结合的原则性技术,将在该领域提供实质性的好处。
信息先验可以施加比硬架构约束编码的偏好更软的偏好。VAE 中的高斯先验就是这样一个例子,尽管它的偏好不受生物信息的引导,这似乎是一个错失的机会。在对 β-内酰胺酶的研究中,研究人员观察到一种类似于蛋白质家族进化所跨越的系统发育树的表示结构。最近旨在强调数据层次结构的双曲线先验可能有助于更清楚地提出这种进化结构。当赋予合适的黎曼度量时,潜在表示可以更好地反映生物学,因此使用相应的几何先验可能很有价值。
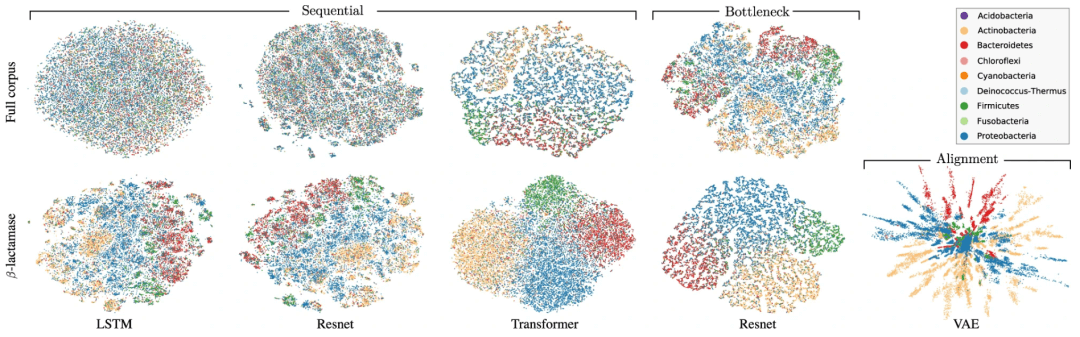
图示:β-内酰胺酶蛋白质家族的潜在嵌入,由分类学在门级进行颜色编码。(来源:论文)

图示:VAE 中 β-内酰胺酶表示之间的最短路径(测地线)。(来源:论文)
适当地分析表示
即使尽了最大的努力将先验知识纳入表征,仍然必须非常小心地解释它们。研究人员强调表示空间中距离的特定示例,并强调看似自然的欧几里德距离具有误导性。现代机器学习方法中编码器和解码器的非线性,意味着表示空间通常是非欧几里得的。
该团队已经证明,通过将观察空间的预期距离以黎曼度量的形式带入表示空间,他们获得的测地线距离与系统发育距离的相关性明显优于通过通常的欧几里得视图可以获得的距离。这是一个令人兴奋的结果,因为黎曼视图带有一组类似于加法和减法的自然算子,因此表示可以在操作上进行。研究人员希望这对蛋白质工程等有价值,因为它提供了一种结合不同蛋白质表达的操作方法。

图示:测地线在潜在空间中提供更稳健和更有意义的距离。(来源:论文)

图示:两个蛋白质序列之间的插值。(来源:论文)
在这项研究中,研究人员仅对变分自编码器的潜在空间进行几何分析,由于其从固定维度潜在空间到固定维度输出空间的平滑映射,因此非常适合。由于他们无法从顺序语言模型中的聚合全局表示进行解码,这一事实阻碍了超出单个蛋白质家族的扩展。
一个自然的问题是,他们提出的瓶颈策略是否可以使这种分析成为可能。如果是这样,它将为定义潜在空间中远程同源物之间有意义的距离提供新的可能性,并可能允许改进蛋白质之间的 GO/EC 注释转移。
最后,几何分析带来了一些与蛋白质无关的含义。它表明,在欧几里得屏幕上将潜在表示绘制为点的常用可视化可能具有高度误导性。因此,科学家认为需要能够忠实反映表示几何的可视化技术。分析还表明,下游预测任务可能会从利用几何结构中获益,尽管标准神经网络架构尚不具备这种能力。
论文链接:https://www.nature.com/articles/s41467-022-29443-w
相关报道:https://phys.org/news/2022-05-machine-potentials-proteins.html

 友情链接
友情链接