








公众号/
编辑 | 萝卜皮
针对核酸大分子,特别是 RNA 的基于结构的药物设计(SBDD)是一个获得动力的研究方向,已经产生了几种 FDA 批准的化合物。与蛋白质类似,SBDD 中 RNA 的关键组成部分之一是正确识别推定候选药物的结合位点。
RNA 具有共同的结构组织,再加上这些分子的动态特性,使得识别小分子的结合位点变得具有挑战性。此外,需要基于结构的方法,因为仅序列信息不考虑核酸大分子的构象可塑性。深度学习有望解决结合位点检测问题,但需要大量的结构数据,与蛋白质相比,这对于核酸来说非常有限。
俄罗斯斯科尔科沃科技学院的研究人员,在一项研究中组建了一组约 2000 个核酸小分子结构,包括约 2500 个结合位点,比以前使用的大 40 倍左右的数据集,并展示了一个基于该结构数据集的深度学习方法 BiteNetN,以检测核酸结构中的结合位点。BiteNetN 使用任意核酸复合物进行操作,显示出最先进的性能,并且有助于分析不同的构象和突变变体。
该研究以「Structure-based deep learning for binding site detection in nucleic acid macromolecules 」为题,于 2021 年 11 月 26 日发布在《NAR Genomics and Bioinformatics》。

RNA 分子在许多细胞过程中至关重要,例如基因调控和细胞信息传递,因此代表了一类有前途的药理靶点。RNA 靶向药物发现活动探索了各种观点,包括 DNA G-四链体稳定剂的设计、靶向核糖开关的抗生素、反义RNA和靶向 RNA 的抗病毒药物等等。扩展可成药基因组的 RNA 靶标,包括那些与「不可成药」蛋白质靶标或非编码 microRNA 相关的靶标,尤其令人感兴趣。
然而,RNA 药物开发存在许多障碍,其中包括与低化学多样性和 RNA 结构的动态特性有关。与蛋白质类似,RNA 分子高度结构化以形成结合位点,小分子可以通过这些位点调节它们。因此,需要高效的、结构特异性的 RNA 小分子配体结合位点检测器来推进 RNA 靶向药物的发现。
「例如,核酸 DNA 和 RNA 可以参与信号传递,我们可以针对它们所参与的信号传递或任何其他过程。对于不可治疗的蛋白质靶标,例如无序蛋白质或缺乏方便结合位点的蛋白质,这可能是一种有希望的策略。」该研究的首席研究员 Petr Popov 说,「然后还有身体外来的致病性 RNA,例如病毒,如 SARS-CoV-2 或 HIV。」
尽管有大量的蛋白质特异性方法,但用于预测 RNA-小分子相互作用位点的方法数量非常有限,大致可分为基于知识的方法、经验方法和机器学习方法。基于知识的方法,例如 InfoRNA,在已知 RNA-小分子结合位点的数据库中挖掘 RNA 基序。经验方法,例如 Rsite、Rsite2 或 RBind,依赖于 RNA 结构的简单几何特征,并寻找这些特征的极端作为结合位点的指标。
最近,科学家开发了一种机器学习方法 RNAsite;它包含一个随机森林模型,该模型使用计算出的 RNA 的基于结构和基于序列的特征进行操作。使用深度学习有望改进 RNA 结合位点检测器;然而,由于可用的 RNA 结构数量相对较少,它受到了阻碍。
事实上,虽然最近用于蛋白质-小分子或蛋白质-肽结合位点检测的深度学习方法依赖于数千个示例的数据集,但 RNAsite 模型仅在 60 个 RNA-小分子复合物上进行了训练。
在这项研究中,该团队展示了第一个基于结构的深度学习方法来预测核酸-小分子配体结合位点。为了克服小数据集的问题,研究人员考虑了 RNA 和 DNA 复合物、与晶体对称配对形成的交互界面、NMR 模型和数据增强。设计组建了一个包含 2000 个核酸小分子结构的数据集,包括从蛋白质数据库(PDB)检索到的 2500 个结合位点接口。
接下来,研究人员开发了基于体素的核链结构视图,每个体素代表物理空间中的一个 1Å3 立方体,并存储对应于特定类型原子密度的八个通道。然后将体素化的表示馈送到 3D 卷积神经网络,该网络对与结合位点有关的核酸结构中的片段进行评分。获得的基于结构的深度学习模型称为 BiteNetN,可预测结合位点界面中心的坐标、每个中心的概率分数以及结合位点中每个核苷酸的分数。
BiteNetN
数据集
为了训练 BiteNetN 深度学习模型,研究人员构建了一个包含 1933 个核酸-配体复合物的大型数据集,包括不同类型的 1065 个 DNA 和 886 个 RNA 结构(18 个结构同时包含 DNA 和 RNA)。
模型
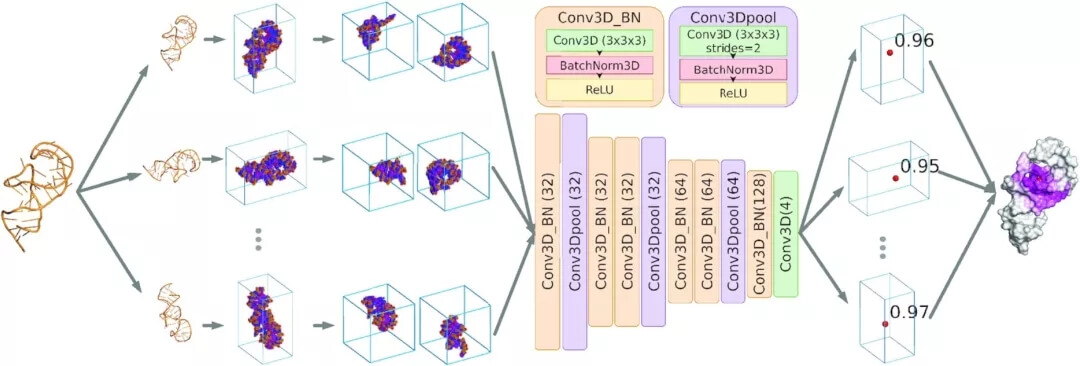
图示:BiteNetN的工作流程。(来源:论文)
研究人员使用 3D CNN 架构在精选的核酸结构上训练 BiteNetN,证明在蛋白质-小分子分子和蛋白质-肽结合位点检测方面表现最佳,上图说明了 BiteNetN 工作流程。
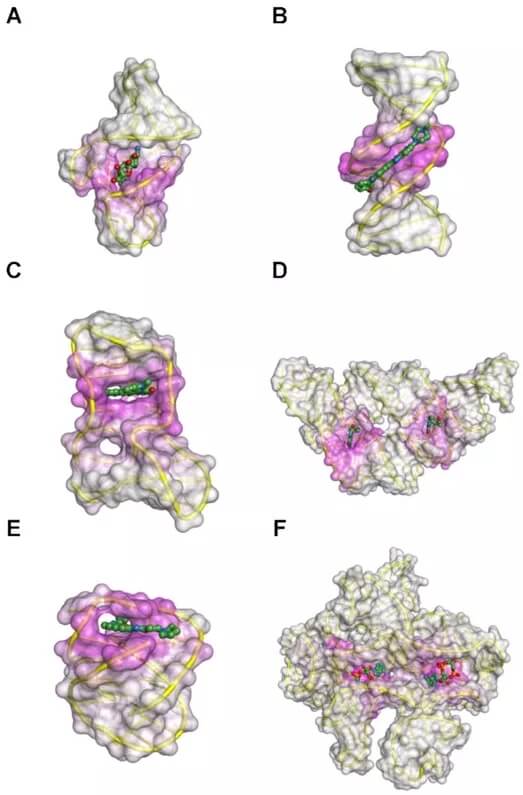
图示:演示 BiteNetN 适用于不同类型的 DNA 或 RNA 结构。(来源:论文)
与其他方法的比较
为了比较 BiteNetN 与其他方法的性能,研究人员获得了四种不同方法的结合位点预测:Rsite、Rsite2、RBind、RNAsite,用于十个测试集。研究人员计算了现有方法的加权 AP、ROC AUC 和 MCC 性能指标,以及在设计的数据集上训练的 10 个 BiteNetN 模型。
「大多数早期的方法只适用于 RNA,特别是单链。我们的方法适用于 DNA 和两条或更多条链。我们甚至可以看到当多个分子纠缠在一起时出现的额外位点。」论文的作者 Igor Kozlovskii 说。
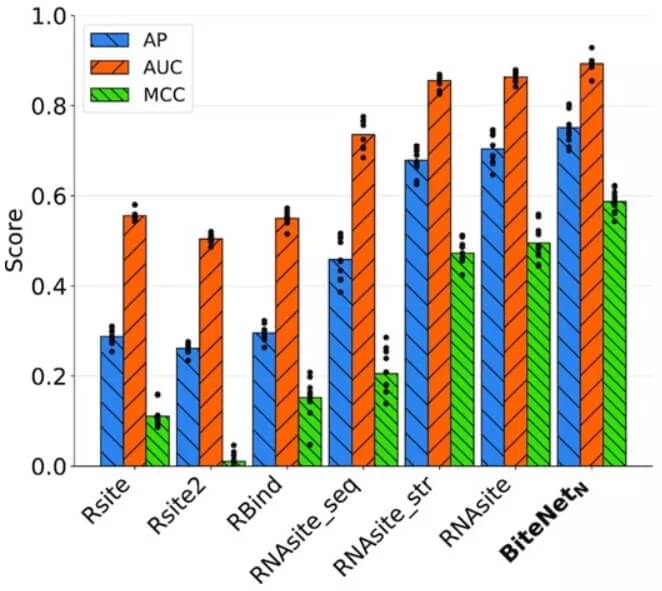
图示:数据集上的加权 AP、ROC AUC 和 MCC 性能指标。(来源:论文)
实例探究
结合位点是大分子的结构和动态特性;因此,预测结合位点的方法应区分具有开放和折叠结合位点的构象,并适用于构象集合的分析。为了证明 BiteNetN 用于相关的核酸配体结合位点检测问题,研究人员测试了 HIV-1 的反式激活反应区域和 ATP 适体。
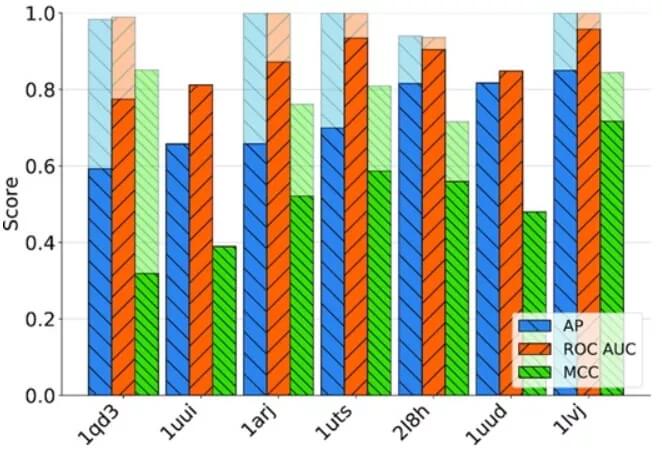
图示:与小分子结合的七种 TAR RNA 结构的 AP 、ROC AUC 和 MCC 性能指标。(来源:论文)
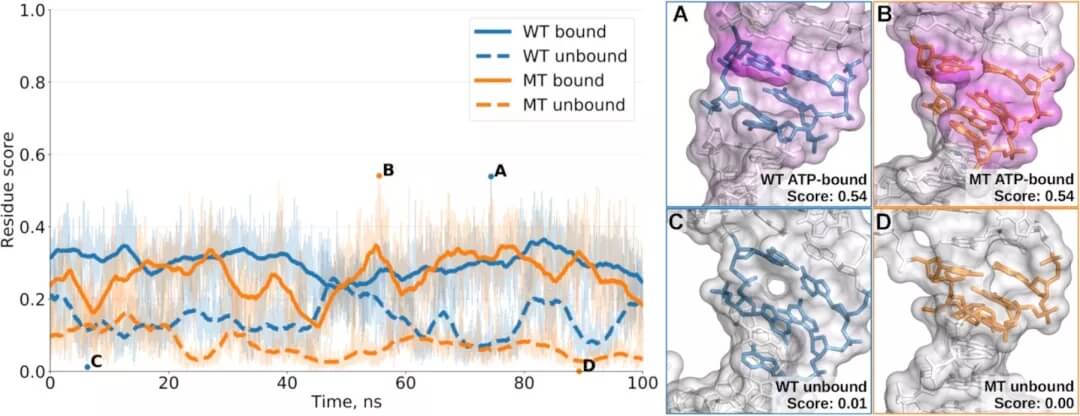
图示:在野生型 ATP 适体及其 G6A 突变体的 ATP 结合和 ATP 未结合 MD 轨迹上计算的结合位点得分。(来源:论文)
总之,该团队想强调的是,核酸结构在原子组成和结构折叠上都不同于蛋白质结构,因此难以直接应用蛋白质结合位点检测方法。在这里,该团队为涵盖各种核苷酸的核酸结构设计了一种特定的类型,适用于 DNA 和 RNA,以及它们的多链复合物。
他们设计的 BiteNetN,在构建的测试集上始终优于其他方法。BiteNetN 具有特定的构象,正如我们通过分析结合小分子的七种不同 HIV-1 TAR RNA 结构所证明的那样。它有助于大规模分析,例如构象集合或突变变异分析,如 ATP 适体案例研究所示。最后,BiteNetN 可以使用 RNA 和 DNA 复合物,包括多条链。
开源链接:https://sites.skoltech.ru/imolecule/tools/bitenet/
论文链接:https://academic.oup.com/nargab/article/3/4/lqab111/6441762#316112271
相关报道:https://phys.org/news/2022-01-artificial-intelligence-rna-dna-sites.html

 友情链接
友情链接