








公众号/ScienceAI(ID:Philosophyai)
编辑/凯霞
原子的 3D 构型决定了材料的属性。从化合物图(chemical graph)、结构式表示中对精确的平衡结构、所有原子的 3D 坐标进行定量预测,是一项具有挑战性且计算成本高的任务,几乎是每个计算化学工作流程的开始。
维也纳大学的研究人员开发了一种新的基于机器学习模型,以简化昂贵的计算,可直接从「图」中预测结构。
该研究于 7 月 22 日以《Machine learning based energy-free structure predictions of molecules, transition states, and solids》为题发表在《Nature Communications》杂志上。

从分子图预测 3D 结构是与自然科学的许多分支相关的普遍挑战。所有原子的元素信息和 3D 坐标定义了系统的电子哈密顿量,从而可以将所有相关的可观测值估计为电子薛定谔方程的近似解的期望值。
在大多数计算高通量筛选活动中,只寻求最稳定的构型。根据估计材料稳定性时所采用近似值的复杂程度,计算单个结构的计算成本可能从几分钟到几小时甚至几天不等。鉴于化合物空间的广阔,所有可能的化合物(估计超过 1,060)所占据的空间,这种成本与质量的权衡是该领域的主要瓶颈。
最近的生成式机器学习发展可能带来了希望。然而,不幸的是,据我们所知,它们尚未用于解决 3D 结构预测问题。
速度提高超一百万倍
现在,由 Anatole von Lilienfeld 领导的研究团队从不同的角度解决了这个问题,开发了一种利用数据并普遍适用于任何类型化学的新方法—— Graph-To-Structure(G2S)。G2S 使用高质量的量子化学数据来训练机器学习模型,该模型能够预测未知化合物分子图的新 3D 结构。
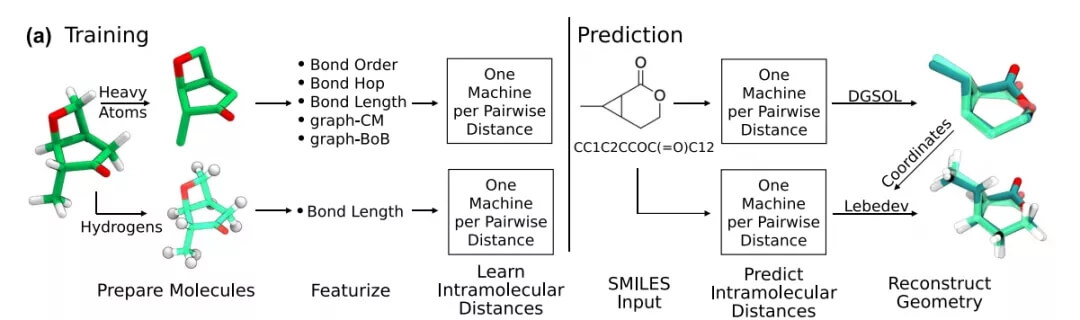
图示:G2S 工作流程。(来源:论文)
为了简单起见,依靠 G2S 的核岭回归 (KRR) 来预测样本外分子或固体的单个原子构型的成对距离矩阵中的所有元素。从成对距离矩阵中,可以轻松地重新创建原子坐标。作为查询输入,G2S 只需要基于键网络和化学计量的信息。利用不含构象异构体的数据集之间的相关性,G2S 学习从化学图到训练数据集中记录的结构最小值的直接映射,从而绕过基于能量的构象搜索和松弛的计算要求过程 。
这种将分子图直接映射到特定 3D 构型的方法,使模型能够有效地绕过任何形式的能量最小化,与传统方法相比,速度提高超一百万倍。「产生高质量结构的可能性不仅加速了高通量分子设计,而且加速了日常工作流程,」该研究的第一作者在 Nature Communications 上说道。「可靠地生成 3D 结构,即使是奇异的化学反应,例如开壳系统或过渡态,是原子模拟中最困难的任务之一。」
进一步的研究结果表明,生成的结构可以直接用作后续基于机器学习的属性预测模型评估的输入,从而以一种更严格、更有效的方式将分子图与结构相关的属性连接起来。
分析和局限性
机器学习预测的分析对于更好地理解 G2S 模型至关重要。研究发现预测的距离分布与各自的参考分布基本重叠。小的偏差表明 G2S 略微高估了共价键的长度,并低估了与第三个邻位的距离。很难辨别第二个邻位的密度差异。
此外,G2S 的学习能力从根本上还没有发挥其全部潜力。准确性的提高将使距离几何问题的解决方案不那么模糊,因此会导致更少的构象异构体/非对映异构体错误分类的情况。
研究人员还尝试构建 Z 矩阵中条目的机器学习模型。然而,基于 Z 矩阵的预测并没有改善基于距离矩阵的模型估计

图示:C7NOH11 结构异构体的 G2S 距离预测分析。(来源:论文)
研究人员表示:「考虑到化学空间的大小和复杂性,一个万能的解决方案只会产生一个更大的模型。从这个意义上说,我们认为 G2S 已经适应了某些感兴趣的化学子空间,并且可以在该领域得到很好的利用,这也是一个重要的优势。未来的工作可以处理粗粒度模拟的应用,玻尔兹曼平均或扩展到预测更多过渡态几何。」
论文链接:https://www.nature.com/articles/s41467-021-24525-7
参考内容:https://phys.org/news/2021-07-chemical-graphs.html

 友情链接
友情链接